

 RNA結合蛋白(RNA binding protiens)結合RNA調控基因表達,與RNA相互作用是聯系多組學的橋梁。從表觀遺傳到蛋白代謝,RNA蛋白互作的研究是高分文章必不可少的。產品介紹經典案例結果展示送樣建議
RNA結合蛋白(RNA binding protiens)結合RNA調控基因表達,與RNA相互作用是聯系多組學的橋梁。從表觀遺傳到蛋白代謝,RNA蛋白互作的研究是高分文章必不可少的。產品介紹經典案例結果展示送樣建議RNA結合蛋白免疫共沉淀測序(RIP-seq)
—全轉錄組范圍揭示RNA與蛋白互作技術
RNA結合蛋白(RNA binding protiens)結合RNA調控基因表達,與RNA相互作用是聯系多組學的橋梁。從表觀遺傳到蛋白代謝,RNA蛋白互作的研究是高分文章必不可少的。RIP-seq(RNA Binding Protein Immunoprecipitation Assay Sequencing,RNA 結合蛋白免疫沉淀測序)通過特定抗體把相應的RNA-蛋白復合物富集沉淀下來,經過分離純化就可以對結合該區域RNA序列進行高通量測序,在轉錄組范圍內鑒定與蛋白結合的區域,是了解轉錄后調控網絡的有力工具。為多組學助力機制研究,艾斯基因引入RIP-seq這一研究轉錄后動態調控技術,精確定位RNA與蛋白互作,助力廣大科研工作者發表高分SCI文章!
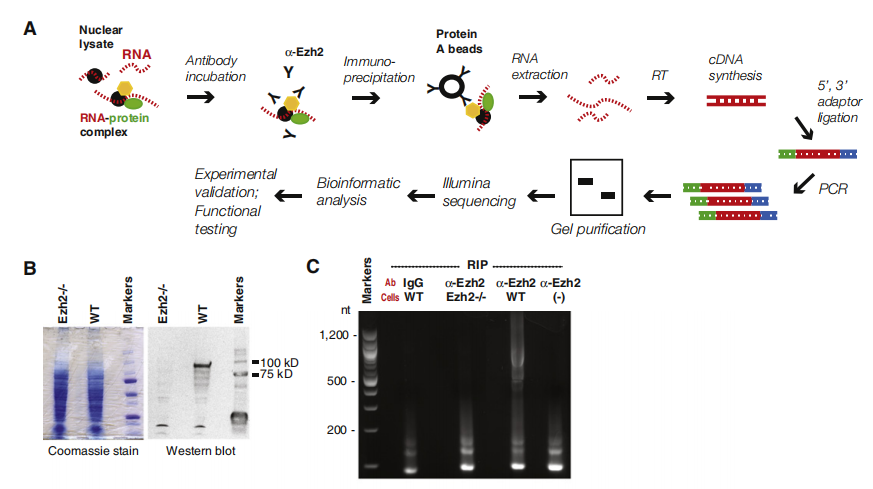
RIP-seq實驗原理圖
艾斯優勢
項目經驗豐富
已經完成人、動植物等幾十個物種, 5000+例樣本項目經驗
專業生信分析團隊
針對多組學測序,艾斯搭建多組學關聯分析流程,多維度助力科研機制研究
高效自動化流程
自動化樣本處理、建庫及分析流程,保障高效周期,40天極速交付
高水平文章經驗
團隊已經發表Genome Biol、Nat Commun、Cell Res等高水平SCI文章
樣本類型和送樣要求
樣本類型
送樣建議
新鮮動物組織
>50 mg
新鮮植物組織
>300 mg
細胞樣本
>5×106個
備注:詳細送樣要求參考 RIP-seq送樣要求2024
數據信息分析
ATAC-seq
分析內容
備注
標準分析
1、測序數據質量評估
過濾掉低質量數據,保證數據質量
2、與參考基因組比對
比對率和覆蓋度分析
3、基因組 Peak 分析
全轉錄組范圍RBP結合RNA的Peak特征
4、Peak關聯基因的功能分析
Peak所關聯基因的GO和KEGG功能富集
5、組間差異Peak分析
尋找差異Peak及注釋
6、組間差異Peak功能分析
差異Peak的GO,KEGG功能富集分析
關聯分析
7、甲基化組學關聯分析
不同甲基化水平基因、DMR的Peak信號分布等
8、轉錄組關聯分析
不同表達水平基因、DEG的Peak信號分布等
9、其它定制化分析
結合課題背景亮點挖掘
在研究中大多數技術都不是單一使用,RIP-seq也不例外,它會搭配其他組學一起對研究的深度和廣度進行擴充。RIP-seq可以與多種技術聯合分析,RNA-seq、m6A-seq等。
案例分析1:m6A識別蛋白YTHDF1在低氧適應和非小細胞肺癌發生發展中的重要功能
YTHDF1 links hypoxia adaptation and non-small cell lung cancer progression. Nat Commun 10, 4892 (2019)..
研究內容:研究發現YTHDF1作為m6A修飾后的RNA結合蛋白家族成員之一,在高原家養哺乳動物中低表達,而在正常肺上皮細胞中抑制其表達可以抵抗低氧誘導的細胞凋亡。深入研究發現,YTHDF1在非小細胞肺癌腫瘤組織和細胞系中均高表達,其在常氧條件下通過加速CDK2和CDK4等細胞周期蛋白的表達來促進腫瘤細胞的增殖;而在鉑類藥物為主的化療壓力環境下,由于YTHDF1高表達促進了Keap1蛋白的表達,導致Nrf2轉錄因子的迅速降解和下游耐藥基因AKR1C1的沉默,因此腫瘤患者對化療更敏感且總生存時間更長
研究技術:RIP-seq、m6A-seq、RNA-seq
研究總結:聯合運用了RIP-seq和m6A-seq,鑒定出細胞周期因子CDK2和CDK4的m6A修飾發生了變化,且被YTHDF1所識別和結合。CDK2和CDK4表達的紊亂也導致細胞周期的紊亂,使得癌細胞增殖加速
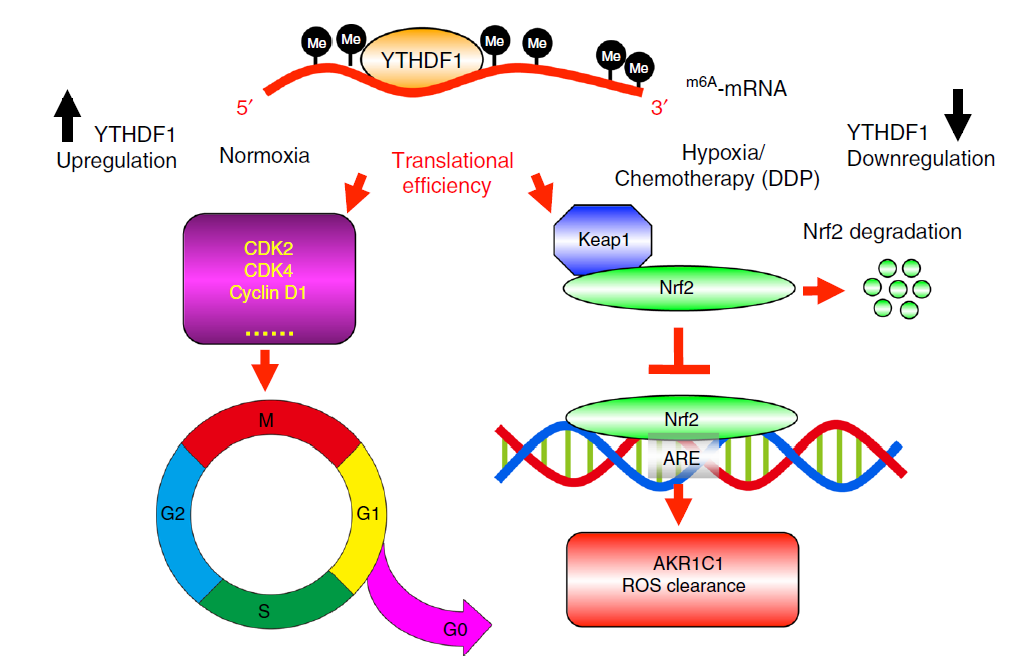
圖1 m6A-seq和 RIP-seq揭示YTHDF1調控模型
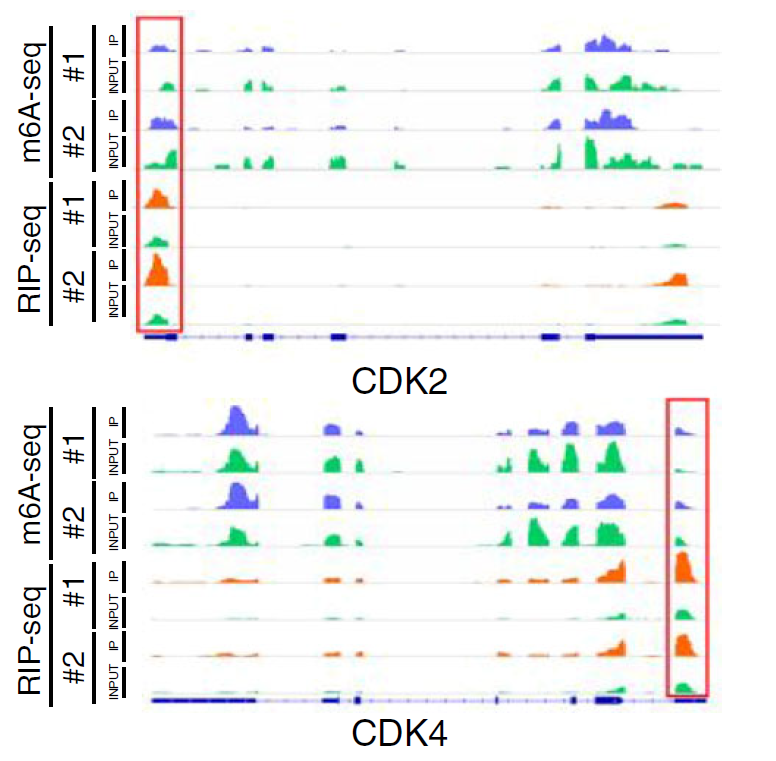
圖2 IGV圖展示CDK2和CDK4的RNA結合蛋白富集差異和m6A修飾差異
案例內容
結果內容
建議內容
產品介紹經典案例結果展示送樣建議RNA結合蛋白免疫共沉淀測序(RIP-seq)
—全轉錄組范圍揭示RNA與蛋白互作技術
RNA結合蛋白(RNA binding protiens)結合RNA調控基因表達,與RNA相互作用是聯系多組學的橋梁。從表觀遺傳到蛋白代謝,RNA蛋白互作的研究是高分文章必不可少的。RIP-seq(RNA Binding Protein Immunoprecipitation Assay Sequencing,RNA 結合蛋白免疫沉淀測序)通過特定抗體把相應的RNA-蛋白復合物富集沉淀下來,經過分離純化就可以對結合該區域RNA序列進行高通量測序,在轉錄組范圍內鑒定與蛋白結合的區域,是了解轉錄后調控網絡的有力工具。為多組學助力機制研究,艾斯基因引入RIP-seq這一研究轉錄后動態調控技術,精確定位RNA與蛋白互作,助力廣大科研工作者發表高分SCI文章!
RIP-seq實驗原理圖
艾斯優勢
項目經驗豐富
已經完成人、動植物等幾十個物種, 5000+例樣本項目經驗
專業生信分析團隊
針對多組學測序,艾斯搭建多組學關聯分析流程,多維度助力科研機制研究
高效自動化流程
自動化樣本處理、建庫及分析流程,保障高效周期,40天極速交付
高水平文章經驗
團隊已經發表Genome Biol、Nat Commun、Cell Res等高水平SCI文章
樣本類型和送樣要求
樣本類型
送樣建議
新鮮動物組織
>50 mg
新鮮植物組織
>300 mg
細胞樣本
>5×106個
備注:詳細送樣要求參考 RIP-seq送樣要求2024
數據信息分析
ATAC-seq
分析內容
備注
標準分析
1、測序數據質量評估
過濾掉低質量數據,保證數據質量
2、與參考基因組比對
比對率和覆蓋度分析
3、基因組 Peak 分析
全轉錄組范圍RBP結合RNA的Peak特征
4、Peak關聯基因的功能分析
Peak所關聯基因的GO和KEGG功能富集
5、組間差異Peak分析
尋找差異Peak及注釋
6、組間差異Peak功能分析
差異Peak的GO,KEGG功能富集分析
關聯分析
7、甲基化組學關聯分析
不同甲基化水平基因、DMR的Peak信號分布等
8、轉錄組關聯分析
不同表達水平基因、DEG的Peak信號分布等
9、其它定制化分析
結合課題背景亮點挖掘
在研究中大多數技術都不是單一使用,RIP-seq也不例外,它會搭配其他組學一起對研究的深度和廣度進行擴充。RIP-seq可以與多種技術聯合分析,RNA-seq、m6A-seq等。
案例分析1:m6A識別蛋白YTHDF1在低氧適應和非小細胞肺癌發生發展中的重要功能
YTHDF1 links hypoxia adaptation and non-small cell lung cancer progression. Nat Commun 10, 4892 (2019)..
研究內容:研究發現YTHDF1作為m6A修飾后的RNA結合蛋白家族成員之一,在高原家養哺乳動物中低表達,而在正常肺上皮細胞中抑制其表達可以抵抗低氧誘導的細胞凋亡。深入研究發現,YTHDF1在非小細胞肺癌腫瘤組織和細胞系中均高表達,其在常氧條件下通過加速CDK2和CDK4等細胞周期蛋白的表達來促進腫瘤細胞的增殖;而在鉑類藥物為主的化療壓力環境下,由于YTHDF1高表達促進了Keap1蛋白的表達,導致Nrf2轉錄因子的迅速降解和下游耐藥基因AKR1C1的沉默,因此腫瘤患者對化療更敏感且總生存時間更長
研究技術:RIP-seq、m6A-seq、RNA-seq
研究總結:聯合運用了RIP-seq和m6A-seq,鑒定出細胞周期因子CDK2和CDK4的m6A修飾發生了變化,且被YTHDF1所識別和結合。CDK2和CDK4表達的紊亂也導致細胞周期的紊亂,使得癌細胞增殖加速
圖1 m6A-seq和 RIP-seq揭示YTHDF1調控模型
圖2 IGV圖展示CDK2和CDK4的RNA結合蛋白富集差異和m6A修飾差異
案例內容
結果內容
建議內容
在線留言
相關產品
在線搜索取消
清空記錄
歷史記錄
清空記錄
歷史記錄
深圳市艾斯基因科技有限公司專注表觀組學技術創新及臨床醫學轉化應用15年
產品中心深圳市艾斯基因科技有限公司關注我們 粵ICP備16072881號-3 粵ICP備16072881號-4版權所有 ? 深圳市艾斯基因科技有限公司粵ICP備16072881號-1
粵ICP備16072881號-3 粵ICP備16072881號-4版權所有 ? 深圳市艾斯基因科技有限公司粵ICP備16072881號-1 選擇區號
選擇區號
 瀏覽器自帶分享功能也很好用哦~?
瀏覽器自帶分享功能也很好用哦~?復制成功
×
 瀏覽器自帶分享功能也很好用哦~
瀏覽器自帶分享功能也很好用哦~

